![]()
Part 1 Principles
1. Fluorescence microscope
2. Filterset
in FL-Mic
3. How concocal differs?
4.
What is confocal?
5.
Resolution in confocal
6. Optical
sectioning
7. Confocal image formation
and
time resolution
8. SNR in
confocal
9.
Variations of confocal
microscope
10. Special features from
Leica sp2 confocal
![]()
Part 2
Application
1. Introduction
2.
Tomographic view
(Microscopical CT)
3. Three-D reconstruction
4. Thick specimen
5. Physiological study
6.
Fluorescence detecting
General
consideration
Multi-channel detecting
Background correction
Cross-talk correction
Cross excitation
Cross emission
Unwanted FRET
![]()
Part
3 Operation and
Optimization
1.
Getting started
2. Settings in detail
Laser line
selection
Laser intensity and
AOTF control
Beam
splitter
PMT gain and offset
Scan
speed
Scan format, Zoom
and Resolution
Frame average, and
Frame accumulation
Pinhole and Z-resolution
Emission collecting rang
and Sequential scan
![]()
When Do
you need confocal?
FAQ
Are
you abusing
confocal?
Confocal Microscopy tutorial
FAQ
![]()
Q1. When do I need confocal
microscope for my fluorescence specimen?
A1: see here.
Q2. Why my specimen looks
different under confocal compared to under conventional microscope?
A2: That is quite normal in confocal microscopy. Due
to the Optical sectioning effect of confocal, you see only one layer of your
specimen under confocal. The thickness of that layer under confocal, depending
on the pinhole size, laser wavelength and objective NA you choose, is generally
at range between 500-800 nm. But your specimen, even if it is a monolayer of
cultured cell, has total thickness of 10-20
µm. What you see is only 1/20 or less of
your cell.
In conventional wide field microscope, you see an overall overlapped image from
top to bottom.
Unless your cell or specimen is homogeneous in size, shape, or in internal
structure along its whole z-direction, you should see some difference.
But if you try to roll the focus through the whole z-axial, you can encounter
all the structures you see under conventional microscope, thought they are
not necessarily in the same plane.
Q3. Why the picture looks even worse under confocal compared to picture under
conventional FL-Mic?
A3: Yes, that is a common phenomena or a problem
of confocal. Confocal effects comes from pinhole and its massive rejecting
out-of-focal-plane signal. The total signal reaches PMT detecter is less than
1/20 of the total signal as mentioned in A2 above. The reduced signal
influx, leads to not only weakened intensity, but also deteriorated SNR (Signal
to Noise Ratio) according to the formula
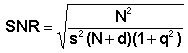 , see section
SNR in confocal. The weakened signal can be
amplified by increasing PMT gain, the deteriorated SNR, however, can not be
compensated by PMT amplification at all. It has to be compensated by using
approaches which increase signal influx such as: more average or accumulation,
lower scan format for bigger pixel size, slower scan speed, bigger pinhole,
etc.. (See related section in this tutorial). If all these fail, that means your
specimen is not suitable for confocal. Your specimen does not have enough signal
intensity, or more important, enough SNR to tolerate the massive signal rejecting
from confocal.
, see section
SNR in confocal. The weakened signal can be
amplified by increasing PMT gain, the deteriorated SNR, however, can not be
compensated by PMT amplification at all. It has to be compensated by using
approaches which increase signal influx such as: more average or accumulation,
lower scan format for bigger pixel size, slower scan speed, bigger pinhole,
etc.. (See related section in this tutorial). If all these fail, that means your
specimen is not suitable for confocal. Your specimen does not have enough signal
intensity, or more important, enough SNR to tolerate the massive signal rejecting
from confocal.
Q4. Which laser line I can use if there is no exact
match of laser line with the excitation peak value of my fluorophore?
A4: Check Table 1,
choose laser line exciting your fluorophore at the highest percentage. Usually,
50-80% is very good. If not available, 25%-50% is acceptable, but you
might need higher AOTF, laser power or PMT setting. If there is similar
percentage at different laser lines, lower wavelength usually gives out better
excitation.
![]()
Statement about this web and
tutorial
For problems or questions regarding this web contact
e-mail:
This page was last updated
23.03.2004